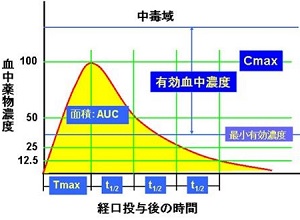
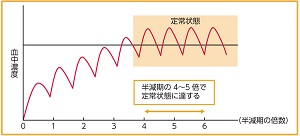
薬が効果を発揮するには、血液を介して目的の組織にまで到達しなければなりません。
錠剤・カプセル剤・散剤などの内服薬であれば、血液に入る前に吸収される必要もあります。
吸収されたとしても、錠剤の形のまま血管の中を流れるわけではありませんので、ここでは、薬がどのような形で吸収や運搬されるのかを紹介します。
イオン化による影響
内服薬の大部分は、水などの液体に溶けた状態で胃を通過して腸に入ります。
腸の粘膜から吸収され、門脈を経て肝臓に入り、そこから全身を巡る血液にまで到達します。
液体に溶けた状態では、イオン結合している薬は一部がイオン化して、解離型と呼ばれる形になります。
イオン化していない形は非解離型と呼びます。
水に溶かした場合の非解離型と解離型の割合は解離定数と言う数値で表され、薬によって決まった割合です。
しかし、非解離型と解離型の割合は常に一定というわけではなく、水ではない液体に溶かした場合には、その液体のphによって変化します。
解離型となった場合は、酸性物質は溶液中でH+を増やす方向に働き、アルカリ性(塩基性)物質はOH-を増やす方向に働きます。
胃の中は胃酸によって強い酸性の状態にあり、ここに薬を入れた場合を考えてみます。
体内を含めて自然界では偏った状況を嫌い、安定のために酸性やアルカリ性も中和する方向に動こうとします。
酸性の液体には元々H+を多く含んでおり、この中に酸性物質が入ると更にH+が増えることになります。
よって、できるだけH+を増やさない方向、つまり非解離型←解離型に変化します。
アルカリ性(塩基性)物質の場合は逆で、OH-によってH+が打ち消されますので、OH-を増やす非解離型→解離型に変化します。
つまり、酸性溶液の中では、水の場合と比べて、酸性物質は非解離型が多くなり、アルカリ性(塩基性)物質は解離型が多くなるわけです。
もちろん、アルカリ性(塩基性)溶液の中では逆になります。
phが1違うと濃度は10倍もの違いになりますので、非解離型と解離型の割合にも大きな変化があります。
ちなみに、解離型はイオン型とも言い、右肩に+や-が付く電荷を帯びた形で、液体中やプラズマ中でないと存在しません。(もしも空気中で解離型となっても、周囲が電荷を打ち消して、すぐに非解離型となります)
吸収する腸の粘膜に話を移します。
水溶性・脂溶性の所でも紹介しましたが、生体膜は疎水構造で、脂溶性物質は通過しやすく水溶性物質は通過しにくい性質があります。(単純拡散の場合で、エネルギーを使って能動輸送する場合は通過します)
正確な表現ではないのですが、解離型を水溶性・脂溶性で区分すると、超が付くほどの水溶性状態です。
つまり、解離型は生体膜をほとんど通過できません。
生体膜の通過は、腸粘膜の吸収だけでなく、血管に入る時や目的組織に移動する時も、さらには排泄される時にも障壁となります。
以上から、「吸収や排泄されるのは非解離型」・「効果を発揮するのも非解離型」になります。
血漿蛋白との結合による影響
血液中に入った薬は、一部がアルブミンという血漿蛋白と結合した状態で循環します。
アルブミンと結合した形を結合型と言い、結合していないものを遊離型と言います。
分子量で比較するとアルブミンは薬の100倍近くある大きさです。
生体膜はこれほど巨大なものを通過できませんので、結合中の薬は組織移行や代謝・排泄もされない一種の隠遁状態になります。
遊離型を徒歩で移動しているとすれば、結合型はバスに乗って移動しているような状態です。(ただし、移動速度は同じですが)
徒歩の場合はコンビニなどへ自由に出入りできますが、バスに乗った状態は一度降車しないとどこへも入ることができず、存在していないのと同じになります。
よって、「移行や排泄されるのは遊離型」・「効果発揮するのも遊離型」になります。
正確に言えば、遊離型の一部は解離型になりますので、効果発揮するのは非解離型の中で遊離型であるものです。
徒歩とバス乗車の割合は薬によってほぼ一定で、遊離型が少なくなると結合型から補充されます。
血漿蛋白質との結合は、新生児黄疸にも関連しています。
胎児期はヘモグロビンが多く、新生児となり、分解されることでビリルビンが多量に生じます。
しかし、肝臓の代謝機能が不十分であるために、血液中にたくさん残った状態になります。
血液中のビリルビンもアルブミンと結合していれば、無色・無害なのですが、新生児にはまだアルブミンが少ないために、遊離型として循環する割合が多くなります。
この結果として生じるのが新生児黄疸です。