| H O M E | |
| 地球と生命の歴史 | |
| 人類進化の歴史 | |
| 文明の誕生へ | |
| 世界史縮尺年表 | |
| 人名歴史年表 | |
| 縮尺:人類史年表 | |
| ネアンデルタール人 |
  |
出アフリカした現生人類はネアンデルタール人と交雑していた(河合信和氏「ヒトの進化七〇〇万年史」p.228~から) 現生人類がヨーロッパに進出した1万年以上後の2万8000年前頃にネアンデルタール人は絶滅したが、これまで考えられていた推定と異なり、両者の間にわずかに交雑があったらしいことが分かった。ドイツ、マックス・プランク進化人類学研究所などの研究チームがネアンデルタール人染色体ゲノムを解読した結果を米科学誌「サイエンス」の2010年5月7日号で報告した。 チームが用いたネアンデルタール人試料は、3.8万年前のクロアチア、ヴィンディヤ洞窟出土の女性3個体化石で、そこから骨片約400ミリグラムを採取し、4年がかりでゲノムの約60%を解読した。その結果を、フランス、中国、パプアニューギニア、アフリカ南部と同西部5人の現代人ゲノムと比較したところ、おおもとのネアンデルタール人祖先が分岐したアフリカ人よりも、それ以外の3地域の現代人の方がネアンデルタール人ゲノムの配列に似ていて、ゲノムの約1~4%はネアンデルタール人由来と推計された。 これまでネアンデルタール人のミトコンドリアDNAの解析は2桁の数に達し、同時に現生人類(ホモ・サピエンス)化石のものとも比較され、両者に交雑のあった証拠がなく、現代人にネアンデルタール人遺伝子は引き継がれていないという説が有力だった。ドイツとアメリカの2つの研究チームが、核DNAの解読を行っていたが、60%という驚異的な解読率により、わずかな交雑の痕跡を捕らえたということになる。ちなみに核DNAは約40億塩基以上、ミトコンドリアDNAはわずか約1万6569塩基で、情報量に大きな違いがある。 研究チームは、現生人類が原郷土のアフリカを出て、10万~5万年前に中東でネアンデルタール人と出会って限定的に交雑し、その後にユーラシア各地に拡散したため、アフリカ以外の現代人でネアンデルタール人由来の遺伝子が検出された、と推定している。 またネアンデルタール人と現代人のゲノムの比較から、現生人類に特徴的な遺伝子として、精神や認知の発達、代謝、頭蓋や胸郭の形成にかかわるものなどが見つかった。 |
| 現生人類のネアンデルタール人との交雑は従来観より新しく6万~5万年前(「河合信和の人類学のブログ」からコピー) |
| ◎08年、西シベリアで発見の4.5万年前の現生人類大腿骨 その時期は、これまで8万年前前後ではなかったかとされているが、イギリスの科学週刊誌『ネイチャー』(2014年)10月23日号に報告された国際研究チームによると、もう少し新しかった可能性が示唆された。 チームは、2008年に西シベリア、ウスチ=イシム近郊にあるイルティシ川の土手で見つかった現生人類の大腿骨化石に残されていたゲノムの塩基配列を解読し、約4万5000年前のものと推定される大腿骨の個体が、ユーラシアに移動した現生人類が西部集団と東部集団に分かれる前か、分かれたのと同じ時期に生きていた男性であることを明らかにした。 またこの個体が、ネアンデルタール人由来のゲノム塩基配列を現代ユーラシア人に見られるのとわずかに少ない程度の量、保有していることも分かった。 |
ReportsTargeted Investigation of the Neandertal Genome It is now possible to perform whole-genome shotgun sequencing as well as capture of specific genomic regions for extinct organisms. However, targeted resequencing of large parts of nuclear genomes has yet to be demonstrated for ancient DNA. Here we show that hybridization capture on microarrays can successfully recover more than a megabase of target regions from Neandertal DNA even in the presence of ~99.8% microbial DNA. Using this approach, we have sequenced ~14,000 protein-coding positions inferred to have changed on the human lineage since the last common ancestor shared with chimpanzees. By generating the sequence of one Neandertal and 50 present-day humans at these positions, we have identified 88 amino acid substitutions that have become fixed in humans since our divergence from the Neandertals.
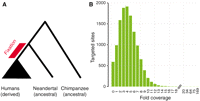
1 Max Planck Institute for Evolutionary Anthropology, D-04103 Leipzig, Germany. 2 Watson School of Biological Sciences, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA. 3 Howard Hughes Medical Institute, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA. 4 Division of Biological Sciences, University of Montana, Missoula, MT 59812, USA. 5 Department of Biology, Emory University, Atlanta, GA 30322, USA. 6 Agilent Technologies, Life Sciences Group, Santa Clara, CA 95051, USA. 7 Área de Prehistoria, Departamento de Historia, Universidad de Oviedo, Oviedo, Spain. 8 Departamento de Paleobiología, Museo Nacional de Ciencias Naturales, Consejo Superior de Investigaciones Científicas, Madrid, Spain. * These authors contributed equally to this work. ↑ 10 Present address: Department of Molecular and Cell Biology, Center for Systems Biology, University of Texas at Dallas, Richardson, TX 75080, USA. ↑ 11 Deceased. ↑ The fossil record provides a rough chronological overview of the major phenotypic changes during human evolution. However, the underlying genetic bases for most of these events remain elusive. This is partly because it is not known when most human-specific genetic changes, identified from genome comparisons to living relatives, occurred during the ~6.5 million years since the separation of the human and chimpanzee evolutionary lineages. However, shotgun sequencing of the Neandertal, a human form whose ancestors split from modern human ancestors 270,000 to 440,000 years ago, has been performed to ~1.3-fold coverage of the entire genome (1). Comparison of Neandertal and present-day human genomes can reveal information about whether genetic changes occurred before or after the ancestral population split of modern humans and Neandertals. However, low-coverage whole-genome shotgun sequencing inevitably leaves a substantial proportion of the genome uncovered. Although deeper shotgun sequencing of one or a few individuals may produce higher coverage across the whole genome, simple shotgun approaches cannot economically retrieve specific loci from multiple individuals, both due to the size of the mammalian genome per se and to the very high proportion (up to 99.9%) of microbial DNA in the vast majority of ancient tissue remains, with the exception of some instances of preservation in permafrost (2, 3). Primer extension capture can isolate specific DNA sequences from multiple Neandertal individuals (4). However, although useful for capture of small target regions such as mitochondrial DNA (mtDNA) (4, 5), this method is unlikely to be scalable up to megabase target regions, ruling out experiments such as the retrieval of exomes, large chromosomal regions, or validation of sites of interest identified in the low-coverage shotgun genome data. Because microarrays can carry hundreds of thousands of probes, we investigated the use of massively parallel hybridization capture on glass slide microarrays (6, 7) on Neandertal DNA at thousands of genomic positions where nucleotide substitutions changing amino acids (nonsynonymous substitutions) have occurred on the human lineage since its split from chimpanzees. For any substitution that is fixed, i.e., occurs in all present-day humans, it is currently impossible to judge how long ago either the original mutation or the subsequent fixation event occurred. However, by ascertaining the Neandertal state at these positions, we can separate fixed substitutions into two classes: (i) sites where a Neandertal carries the derived state, which indicates that the substitution must have occurred before the population split of modern humans and Neandertals; and (ii) sites where a Neandertal is ancestral, which indicates that fixation of a substitution in modern humans occurred after the population split with Neandertals (Fig. 1A).
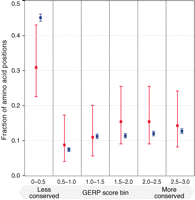
To identify substitutions that occurred on the human lineage since the ancestral split with chimpanzee, we aligned human, chimpanzee, and orangutan protein sequence for all orthologous proteins in HomoloGene (8, 9). Comparison of these three species allowed us to assign human/chimpanzee differences to their respective evolutionary lineages. We designed a 1 Million Agilent oligonucleotide array covering, at 3–base pair tiling, all 13,841 nonsynonymous substitutions inferred to have occurred on the human lineage (9). We used this array to capture DNA from a ~49,000-year-old Neandertal bone (Sidrón 1253) from El Sidrón Cave, Spain (10, 11). This bone contains a high amount of Neandertal DNA in absolute terms, but also a high proportion (99.8%) of microbial DNA (4), making it unsuitable for shotgun sequencing. To identify which of the 13,841 substitutions are fixed in present-day humans, we also collected data from 50 individuals from the Human Genome Diversity Panel (12) with the same array design as used for the El Sidrón Neandertal (table S1). The DNA libraries from these individuals were barcoded, pooled, and captured on a single array (13). All captured products were sequenced on the Illumina GAII platform and aligned to the human genome (9). Overall, 37% of the Neandertal sequence reads aligned to the target regions, representing ~190,000-fold target enrichment. We retrieved Neandertal sequence for 13,250 (96%) of the substitutions targeted on the array, with an average coverage of 4.8-fold after filtering for polymerase chain reaction (PCR) duplicates (Fig. 1B). We considered a Neandertal position ancestral if all overlapping reads matched the chimpanzee state and derived if all reads carried the modern human state or if we found a mixture of derived and third-state reads, disregarding positions that carried only a third state or positions where Neandertal reads were found both in the ancestral and in the derived state. From each present-day individual, a total of 25% (23 to 27%) of reads aligned to the target regions. In each individual, we retrieved on average 98% (97 to 99%) of targeted positions and had on average coverage of 10-fold (fig. S1). We estimated genotypes for each individual and considered a position to be fixed derived if it was homozygous and derived in all humans observed, and if data were available for at least 25 individuals (50 chromosomes) (9). We included several additional target regions on the array to assess levels of human DNA contamination, which can frequently affect ancient DNA experiments (14). One such region was the complete human mtDNA, which is known to differ between the Sidrón 1253 Neandertal analyzed here and almost all (99%) present-day humans at 130 positions (4). Even though the array probes were designed to match present-day human mtDNA, 253,549 of the 254,296 (99.71%) fragments that overlapped these 130 positions matched the Neandertal state. We therefore conclude that the vast majority of mtDNA in the Sidrón 1253 library is of Neandertal origin. For a more direct estimate of contamination in the nuclear DNA, we used 46 nucleotide sites on the X chromosome that differ between present-day humans and chimpanzees and that were found to be ancestral in a Neandertal from Croatia (Vindija 33.16) by shotgun sequencing (1), whereas ~1000 present-day humans in the human diversity panel carry a derived state. The Sidrón 1253 individual will obviously not match Vindija 33.16 at all of these sites. However, because Sidrón 1253 is a male (15) and thus carried a single X chromosome, at sites where he does match Vindija 33.16, all reads should carry the ancestral base while apparent heterozygosity will indicate human DNA contamination. By analyzing the consistency of reads overlapping these sites on the X chromosome, we calculated a maximum likelihood estimator of X-chromosomal contamination of 4%, although confidence intervals are large (1 to 12%) due to the small number of relevant positions (9). Another way to estimate contamination across autosomes is to investigate patterns of allele counts. Because at every site an individual is either homozygous derived, homozygous ancestral, or heterozygous, DNA from a single individual will yield at each site either only derived alleles, only ancestral alleles, or a draw with equal chance for either. Contamination from other individuals would cause systematic deviation from these patterns. We thus produced a likelihood model that estimated contamination at the positions recovered from Sidrón 1253, and calculated a 95% upper bound for contamination of 2% (9). From these results we conclude that the Sidrón 1253 data are not substantially affected by human DNA contamination. In total, we determined with high confidence the Neandertal and present-day human state for 10,952 nonsynonymous substitutions. In 10,015 (91.5%) of all cases the Neandertal carries the derived state, whereas in 937 (8.5%) cases the ancestral state was found (fig. S2). Of the positions that are fixed in the derived state in present-day humans, 9525 (87%) are derived in Neandertal, whereas 88 (0.8%) (table S2) are ancestral (fig. S2). In agreement with previous results generated by PCR (15), two substitutions that change amino acids in the gene FOXP2 (16), involved in speech and language (17), are both derived in this Neandertal individual. The 88 recently fixed substitutions occur in 83 genes (tables S2 and S3). We asked if these genes cluster in any group of functionally related genes relative to the genes that were targeted in the capture array (18) (as defined in the Gene Ontology) but found no such groups. We furthermore asked if the 88 substitutions that recently became fixed in humans differ from those that occurred before the divergence from the Neandertal with respect to how evolutionarily conserved the positions in the encoded proteins are (9, 19) (Fig. 2). We found that the 88 recent substitutions tend to affect amino acid positions that are more conserved than the older substitutions (Wilcoxon rank text; P = 0.014). Similarly, the recently fixed substitutions caused more radical amino acid changes with respect to the chemical properties of the amino acids (Wilcoxon rank test; P = 0.04). One possible explanation for these observations is that the effective population size of humans since their separation from the Neandertal lineage has been small, leading to a reduced efficiency of purifying selection, as seen, e.g., in Europeans (20). We also looked for evidence that the recent substitutions may have been fixed by positive selection. One recent substitution occurred in SCML1, a gene involved in spermatogenesis (21) that has been previously proposed as a target of positive selection in humans (22) as well as frequent positive selection in primates (23). However, we found no significant overrepresentation of the 83 genes among candidate genes in three genome-wide scans for positive selection (24) (table S4). Nevertheless, we believe that all of these amino acid substitutions warrant functional studies.
Our results demonstrate that hybridization capture arrays can generate data from genomic target regions of megabase size from ancient DNA samples, even when only ~0.2% of the DNA in a sample stems from the endogenous genome. By generating an average coverage of 4- to 5-fold, errors from sequencing and small amounts of human DNA contamination can be minimized. A further approximately 5-fold reduction of errors was achieved here by the enzymatic removal of uracil residues that are frequent in ancient DNA (25). Because the Sidrón 1253 Neandertal library used for this study has been amplified and effectively immortalized, the same library should be able to provide similar-quality data for any other genomic target region, or even the entire single-copy fraction of the Neandertal genome. Supporting Online Material
www.sciencemag.org/cgi/content/full/328/5979/723/DC1 Materials and Methods Figs. S1 to S4 Tables S1 to S5 References and Notes
Received for publication 8 February 2010. Accepted for publication 1 April 2010. The editors suggest the following Related Resources on Science sites:In Science Magazine
|
Copyright © Libell 2012. All rights reserved.